Weiterführende Informationen und Differentialdiagnostik zur Zertifizierten Kasuistik "Patient mit Exsikkose und unklarer Schluckstörung"
von René Trabold
Inhaltsübersicht
Epikrise des Patienten in unserer Kasuistik
Erläuterungen zur Kasuistik
Das Vorliegen einer tageszeitlich und belastungsabhängig fluktuierenden Bulbärsymptomatik lässt an ein Myasthenie-Syndrom denken. Die Verdachtsdiagnose unterstützend zeigen sich im klinischen Befund eine Ptose, welche durch den Simpson-Test (Ermüdungstest: prolongierter Blick nach oben führt innerhalb einer Minute zur Ptose oder Doppelbildangabe), das Cogan-Zeichen (tonische Aufwärtsbewegung des Oberlides) sowie durch den Eisbeuteltest (Eisbeutel auf dem ptotischen Auge für wenige Minuten führt zur Besserung der Ptose) modulierbar ist. Zusätzlich bestand eine Schwäche im Bereich der Nackenmuskulatur.
Darüber hinaus blieb die cerebrale Bildgebung ohne wegweisenden pathologischen Befund. Die Gabe von einem Acetylcholinesterasehemmer führte (diagnostisch) zur nahezu vollständigen Restitutio und bestätigte somit die Verdachtsdiagnose.
Im weiteren Verlauf bestand nun die Notwendigkeit einer ursächlichen Einordnung des Myasthenie-Syndroms. Dieses Syndrom kommt als häufigstes paraneoplastische Syndrom bei Thymomen vor.
Definition und Klassifikation
Die Myasthenia gravis beruht pathophysiologisch auf einer Störung der neuromuskulären Erregungsübertragung. Bei der häufigsten Form finden sich ursächlich Autoantikörper gegen nikotinische Acetylcholin-Rezeptoren. Es gibt darüber hinaus zahlreiche Subgruppen unterschiedlichster Autoantikörper mit einer hohen Antigendiversität.
Die Inzidenz der Myasthenia gravis liegt bei circa 0,25 bis 2,0 pro 100 000 Einwohnern. Es besteht ein familiär erhöhtes Risiko bei HLA-Trägern (B8 und DR3). Die generalisierte Myasthenia gravis wird je nach Krankheitsbeginn als „early- onset“ vs. „late-onset“ unterschieden. Als Bezugspunkt dient das 45. Lebensjahr. 10 bis 15 Prozent aller Patienten haben ein Thymom.
Die Myasthenia gravis läßt sich einteilen in eine leichte (rein okuläre) bis schwerste (generalisiert; intensiv- und intubationspflichtig) Verlaufsform entsprechend der Klassen I-V (mod. nach Myasthenia Gravis Foundation of America). Sie kann assoziiert sein mit rheumatologischen-vaskulitischen Erkrankungen. Besonders hervorzuheben ist ihr Bezug zu Schilddrüsenfunktionsstörungen (Thyreoditis, endokrine Orbitopathie).
Klinisches Bild - Verlauf
Entsprechend der beteiligten Muskeln beziehungsweise Muskelgruppen findet sich eine belastungsabhängige, im tageszeitlichen Verlauf fluktuierende Muskelschwäche. Zur Einteilung, Verlaufs- und Therapiekontrolle findet der sogenannte Besinger Score (1) seine Anwendung.
Verlauf
Es besteht eine Tendenz zur Generalisierung der Erkrankung. Lediglich 10 bis 20 Prozent der Krankheitsverläufe bleiben auf eine rein okuläre Symptomatik beschränkt. Jede Mitbeteiligung anderer Muskelgruppen wird als generalisiert (unabhängig der Ausprägung) bezeichnet. Der Krankheitsverlauf wird durch äußere Einflussfaktoren wie Infekte, Stress, Medikamente oder hormonelle Faktoren beeinflusst. Zum Beispiel erscheint der Verlauf im 2. und 3. Trimenon einer Schwangerschaft etwas abgemildert.
Praktische Bedeutung hat die stark erhöhte Empfindlichkeit und damit Symptomverstärkung der Myasthenia gegenüber muskelrelaxierenden Substanzen vom Curare-Typ (Dosisanpassung notwendig), Benzodiazepinen und Strukturverwandten sowie einigen Antibiotika (Aminoglykoside, Tetrazykline, Gyrasehemmer, Makrolide und Ketolide). Penicilline gelten zumindest im Niedrigdosisbereich als sicher.
Diagnostik
Neben den erwähnten anamnestischen wie auch klinischen Stigmata erfolgt die Gabe von Cholinesterasehemmern als diagnostisch zielführend (favorisiert: orale Gabe von 30 bis 60 mg Pyridostigmin). Desweiteren erfolgt eine elektrophysiologische Untersuchung mittels supramaximaler repetitiver Reizung interessierender / betroffener Muskelgruppen. Als pathologisch wird ein sogenannter Dekrement der Reizantwortamplitude gewertet. Der Nachweis eines Dekrements gelingt bei generalisierter Verlaufsform im Allgemeinen besser. Als spezielle Untersuchungsmethode gilt die Einzelfasermyographie.
Neben den Standardparametern in der Labordiagnostik wird eine Autoantikörperdiagnostik angeschlossen (Ach-R-AK+ Diversitäten). Die Diagnostik erweitert sich bei negativem Befund beziehungsweise entsprechend der speziellen (differentialdiagnostischen) Fragestellung.
Zur Klärung retrosternaler bzw. thorakaler Auffälligkeiten wird eine Thorax-CT/MRT empfohlen. In der vorliegenden Kasuistik ist im CT (siehe Abbildung) die mediastinale Raumforderung im Sinne eines Thymoms zu erkennen.
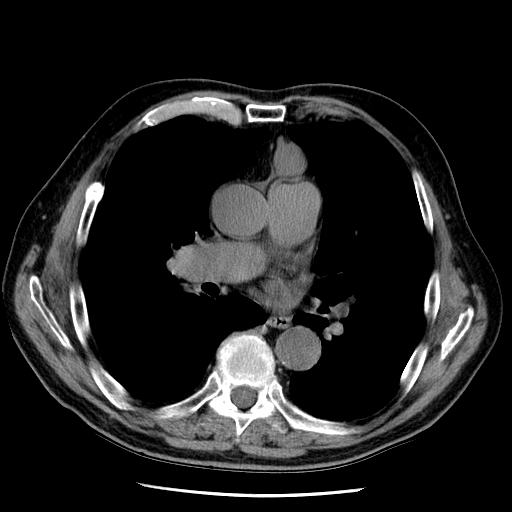
Computertomographie des Thorax. Foto: Benedictus Krankenhaus, Tutzing
Differentialdiagnosen
Die Liste der Differentialdiagnosen umfasst in erster Linie
- sämtliche myopathische und myositische Erkrankungen wie Pathologien an den alpha-Motoneuronen
- Medikamenten-induzierte Phänomene (z. B. bei D-Penicillamin, Chloroquin),
- autoimmunologische Polyneuritiden sowie auch
- Erreger-assoziierte Erkrankungen (z. B. Botulismus) usw.
Erwähnt sei hier auch das Lambert-Eaton-Syndrom mit Bildung paraneoplastischer Antikörper gegen präsynaptische spannungsabhängige Kalziumkanäle.
Therapie
Hinsichtlich therapeutischer Überlegungen entscheidet sich (die Ursache als bekannt vorausgesetzt) eine symptomatische Therapie von einer immunsuppressiven, gegebenenfalls operativ-kurativen.
Die wichtigste symptomatische Therapie stellt die Gabe von Cholinesterasehemmern dar, deren Dosierung sich nach dem klinischen Effekt richtet. Bei nicht ausreichendem Ansprechen auf eine rein symptomatische Behandlung wird eine immunsuppressive Basistherapie eingeleitet. Steroide und Azathioprin sind Basistherapeutika in der Behandlung der Myasthenie. Bei der Behandlung mit Azathioprin ist auf comedikamentöse Dosisanpassungen beziehungsweise Wechselwirkungen zu achten. Auch kann die Bestimmung genetischer Variationen der Thiopurinmethyltransferase (TPMT) hilfreich sein. Bei Therapieresistenz kann unter anderem eine Behandlung mit Rituximab erfolgreich sein.
Myasthene Krise
Die Behandlung einer myasthenen Krise hat auf einer Intensivstation zu erfolgen. Es gilt neben der Vermeidung von Infekten die rasche Wiederherstellung der neuromuskulären Erregungsübertragung sowie die Kupierung der Autoimmunreaktion (z. B. durch Plasmapherese, Immunadsorption, Immunglobuline). Ein krisenhafter Verlauf ist möglich bei Infektkonstellationen, medikamenten-induziert, aber auch bei Elektrolytveränderungen.
Thymektomie
Patienten in der Subgruppe des „ early-onset“ mit generalisierter Myasthenie profitieren von einer Thymektomie, wenn diese innerhalb eines gewissen Zeitraumes durchgeführt wird. Bei fehlendem Nachweis von Autoantikörpern erscheint der Vorteil einer Thymektomie fraglich beziehungsweise nicht gegeben. Wird ein Thymom nachgewiesen, so besteht die Indikation zur Operation. Bei lokal-invasiven Thymomen ist die adjuvante Chemotherapie indiziert.
Epikrise des Patienten in unserer Kasuistik
In der vorliegenden Kasuistik lag klinisch die Verdachtsdiagnose eines Myasthenie-Syndromes beziehungsweise einer Myasthenia gravis nahe. Diese Verdachtsdiagnose wurde bestätigt durch die Gabe von Pyridostigmin mit der hierauf folgenden eindrücklichen Befundbesserung. Die im Rahmen der weiteren Diagnostik durchgeführte CT-Thorax-Aufnahme (siehe Abbildung) zeigte eine Raumforderung im vorderen Mediastinum dem Perikard aufliegend, ohne infiltrative Komponente und entsprach einem Thymom.
Es erfolgte zunächst die optimierte Dosisanpassung des Acetylcholinesterasehemmers (Pyridostigmin) bis zur vollständigen klinischen Beschwerdefreiheit. Nach umfangreicher Diagnostik und Thymombestätigung im CT wurde dieser Tumor thorakoskopisch entfernt. Im kurzzeitigen postinterventionellen Intensivsetting konnte eine drohende Myastheniekrise durch Neostigmin-Gabe (intravenös) Gabe kupiert werden. Bereits am Folgetag nach der Operation war die orale Einnahme von Pyridostigmin wieder möglich und ausreichend.
Wir beschlossen eine Kontrolluntersuchung des Patienten nach rund zwei Monaten mit versuchsweiser Reduktion der Pyridostigmindosis. Eine immunsuppressive Therapie wurde (noch) nicht eingeleitet, zumal nach vollständiger Thymomentfernung eine Restitutio ad integrum wahrscheinlich ist.
Literatur
- Besinger UA, Toyka KV, Heininger K et al. (1981)
- Es gilt allgemein: Angaben, Daten und Zahlen sowie Empfehlungen gemäß der gültigen Leitlinie „Diagnostik und Therapie der Myasthenia gravis und des Lambert-Eaton-Syndroms“ (Entwicklungsstufe S2k)