Weiterführende Informationen und Differentialdiagnostik zur Zertifizierten Kasuistik "Multiple Osteolysen unklarer Genese "
von Laura Pennington, Julia Wagenpfeil, Christoph Sippel und Peter Walger
Inhaltsübersicht
Definition und Ursache des primären Hyperprarathyreoidismus
Pathogenes und klinisches Bild
Allgemein
Im vorliegenden Fall litt die Patientin unter einer Hyperkalzämie bedingt durch einen primären Hyperparathyreoidismus bei einem solitären Nebenschilddrüsenadenom rechts kaudal der Schilddrüse. Durch ihre eingeschränkten finanziellen Ressourcen und die fehlende Inanspruchnahme einer ärztlichen Abklärung in den vier Jahren der Erkrankung, kam es zur Entwicklung des heute in hochentwickelten Industrieländern sehr seltenen Vollbildes der Krankheit mit schweren Osteolysen im Rahmen einer Ostitis fibrosa cystica generalisata von Recklinghausen. Diese waren anfänglich zum Anlass genommen worden, eine maligne Gunderkrankung zu vermuten. Zentraler Laborwert zur Diagnosesicherung war in diesem Fall das Parathormon (PTH intakt), welches bei unklaren Osteolysen immer bestimmt werden sollte.
Definition und Ursache des primären Hyperparathyreoidismus
Der primäre Hyperparathyreoidismus (pHPT) ist definiert als eine vermehrte Sekretion von Parathormon (PTH) durch eine Erkrankung der Nebenschilddrüsen (NSD). Die Prävalenz beträgt circa 1-4/1000 Patienten, wobei Frauen doppelt so häufig, jedoch durchschnittlich später als Männer erkranken (w: 60. - 70. LJ; m: 40. - 50. LJ).
Meist liegen der Erkrankung ein solitäres (80 %) - wie im beschriebenen Fall - oder multiple (5 %) Adenome der NSD zugrunde. Diese entstehen in der Regel durch monoklonale Mutation einer PTH-produzierenden Hauptzelle. Im Gegensatz dazu liegt bei der selteneren ursächlichen Hyperplasie (15 %) eine polyklonale Proliferation der parathyreoidalen Zellen vor. Nur selten (< 1 %) findet sich ein Karzinom (Mutation des Retinoblastom-Suppressorgens) als Auslöser.
Der pHPT tritt bisweilen auch im Rahmen der multiplen endokrinen Neoplasie (MEN) Syndrome auf. Ihnen liegen autosomal dominant vererbte Mutationen des Menin-Gens (MEN1) oder des Ret-Protoonkogens (MEN2A/2B) zugrunde. Daher sollte bei Erstdiagnose eines pHPT immer auch an die Möglichkeit eines MEN-Syndroms gedacht werden! Tabelle 1 gibt eine kurze Übersicht der Syndrome.
| Tabelle 1: Die verschiedenen MEN-Syndrome und ihre Vererbung | |||
|---|---|---|---|
| MEN1 | MEN 2a (70 %) | MEN 2b (10 %) |
Vererbung | Autosomal-dominant (Menin-Gen) | Autosomal-dominant (Ret-Protoonkogen) | Autosomal-dominant (Ret-Protoonkogen) |
Leittumor | Pankreastumor (50 %) (Insulinom/ Gastrinom) | Medulläres Schilddrüsen-Karzinom (100 %) | Medulläres Schilddrüsen-Karzinom |
PLUS | pHPT (95 %) | pHPT (20 %) | Phäochromozytom |
| Hypophysentumore (30 %) | Phäochromozytom (50 %) | Schleimhautneurinome Marfanoider Habitus |
Im vorliegenden Fall wurde vor allem die Möglichkeit eines begleitend vorhandenen medullären Schildrüsenkarzinoms sonographisch und laborchemisch (Calcitonin im Normbereich) ausgeschlossen. CT-morphologisch fanden sich keine Hinweise auf abdominelle Begleitmalignome.
Pathogenese und klinisches Bild
Kalzium liegt im Serum beziehungsweise Plasma zu rund 50 Prozent in seiner freien, ionisierten Form und zu 50 Prozent protein- (v.a. an Albumin) oder komplexgebunden vor. Der Kalziumspiegel ist einer der am präzisesten regulierten physiologischen Parameter. Dabei kommt dem negativen Rückkopplungsmechanismus zwischen Kalzium und PTH die zentrale Rolle zu. Beim pHPT wird jener durch eine kalziumunabhängige autonome PTH-Produktion außer Kraft gesetzt mit der Folge einer Entgleisung des Kalziumspiegels in Form einer Hyperkalzämie. Dies lässt sich auf drei Angriffspunkte von PTH zurückführen.
Erstens kommt es zur vermehrten Freisetzung von Kalzium aus den Knochen. PTH stimuliert zwar Osteoklasten und Osteoblasten, bewirkt jedoch in pathologisch erhöhten Konzentrationen eine Negativierung der Knochenbilanz. Infolge dessen kommt es zu einer fortschreitenden Osteopenie und in ausgeprägten Fällen – wie in dieser Kasuistik - zur Entstehung von Osteolysen im Bereich des Beckens und Hüftknochen (Abbildung 1) sowie subperiostalen Resorptionslakunen sowie zu Akroosteolysen an Händen und Füßen (Abbildungen 2 und 3).
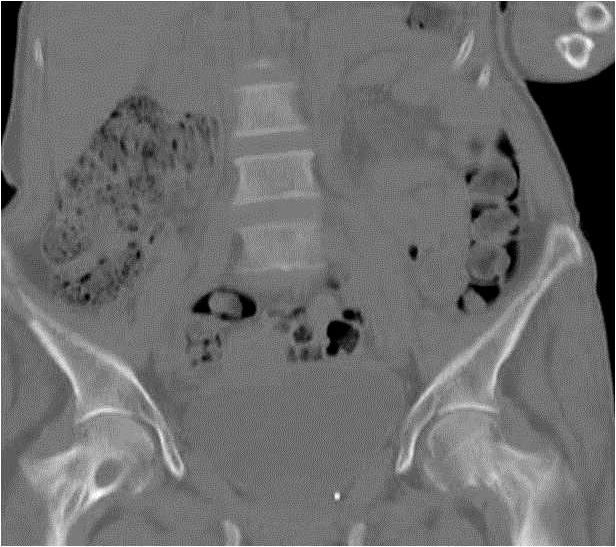
Abbildung 1: CT-LWS-/Beckenknochen und Hüftknochen der Patientin mit Osteolysen
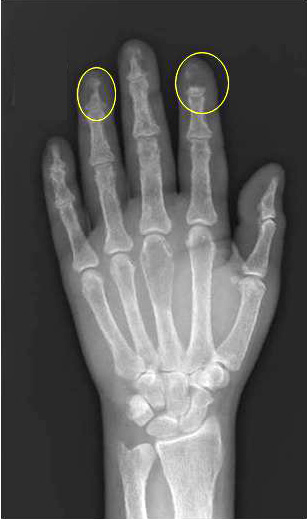
Abbildung 2: Röntgenaufnahme der linken Hand der Patientin mit subperiostalen Resorptionslakunen sowie zu Akroosteolysen (z. B. gelb).
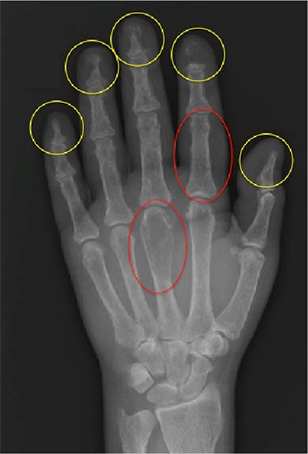
Abbildung 3: Röntgenaufnahme der linken Hand der Patientin mit großen subperiostalen Resorptionszonen (rot) und ausgeprägten Akroosteolysen (gelb).
Abbildungen: Johanniter Krankenhaus Bonn.
Darüber hinaus kann es zu einem typischen „Mattglas-Effekt“ des Schädels (auch „Pfeffer-Salz-Schädel“) kommen mit Erosionen der Lamina dura der Zahnalveolen. Im geschilderten Fall entwickelten sich zudem die heute kaum noch zu beobachtenden „braunen Tumoren“ im Rahmen einer Ostitis fibrosa cystica generalisata von Recklinghausen. Diese entstehen durch bindegewebigen Ersatz abgebauten Knochengewebes (Fibroosteoklasie) und spätere Einblutung in diese Resorptionszysten. Abbildung 4 zeigt einen braunen Tumor im histopathologischen Oberarmpräparat der Patientin in 200-facher Vergrößerung. Klinisches Korrelat dieser ossären Veränderungen sind Glieder- und Rückenschmerzen (50 % der Fälle) sowie pathologische Frakturen.
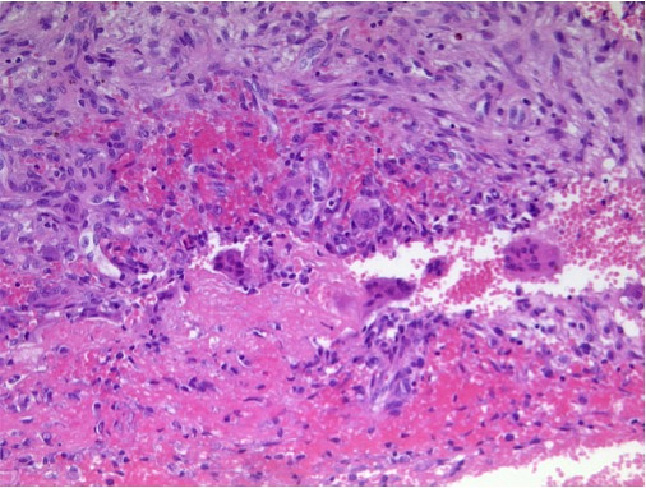
Abbildung 4: Histopathologisches Humerus-Präparat der Patientin in 200-facher Vergrößerung. Es findet sich ein Fibroplastenproliferat mit Hyperämie und reichlich Siderophagen im Sinne eingebluteter Resorptionszysten als Korrelat brauner Tumoren bei pHPT.
Quelle: Institut für Pathologie Prof. Büttner, Uniklinikum Köln
Zweitens fördert PTH die Hydroxylierung von 25-(OH)-Vitamin D in der Niere zu 1,25-(OH)2-Vitamin D und damit die intestinale Rückresorption von Kalzium. Zuletzt führt PTH auch zur vermehrten renalen Rückresorption von Kalzium im distalen und Sekretion von Phosphat im proximalen Tubulus, wodurch die Menge des freien und damit aktiven Kalziums im Blut steigt.
Durch die Verschiebung des Löslichkeitsproduktes sowie die insgesamt durch die Hyperkalzämie erhöhte Kalziurese kommt es vermehrt zu Nephrolithiasis und - prognostisch ungünstiger - selten auch zur Nephrokalzinose (renale Manifestation in 40-50 % der Fälle). Zudem bewirkt der pHPT eine ADH-refraktäre Einschränkung der Urinkonzentrierung mit konsekutiver Polyurie und Polydipsie.
Die Hyperkalzämie bewirkt darüber hinaus eine vermehrte Gastrinsekretion mit resultierenden gastrointestinalen Beschwerden (50 %) wie Appetitlosigkeit und Übelkeit sowie seltener Ulcera ventriculi oder duodeni. Auch unspezifische Symptome wie Meteorismus, Obstipation und Gewichtsabnahme sind häufig. Selten kommt es auch zur Pankreatitis.
Die englische Variante („stones – bones – abdominal moans – psychic groans“) des deutschen Merkspruches „Stein – Bein – Magenpein“ schließt zudem noch mögliche psychische und neuromuskuläre Begleiterscheinungen wie depressive Verstimmungen, Muskelschwäche, -atrophie und rasche Ermüdbarkeit mit ein. Bei fortgeschrittenen Krankheitsausprägungen kann es auch zu schwerwiegenderen psychiatrischen Manifestationen bis hin zur Psychose kommen.
Allerdings sind bis zu 50 Prozent der Patienten bei Diagnosestellung a- oder oligosymptomatisch.
Diagnostik
Der pHPT ist primär eine laborchemische Diagnose anhand des diagnostischen Paares Kalzium und Parathormon. Typischerweise sind beide Parameter beim pHPT erhöht. Bei Knochenmanifestation, welche in circa 50 Prozent der Fälle vorliegt, kann zudem die alkalische Phosphatase erhöht sein. Als Leitfaden gilt, dass eine an drei verschiedenen Tagen gemessene Erhöhung des Serumkalziums > 2,6 mmol/l sowie des PTH intakt bei normaler Nierenfunktion und normalem Gesamteiweiß mit > 95 % Sicherheit für einen pHPT spricht!
Meist ist die Hyperkalzämie auslösend für eine weitere Abklärung. Zur Differenzierung zwischen primärem, sekundärem und tertiärem HPT sollten immer PTH intakt und 25-(OH)-Vitamin D bestimmt werden. Bei jüngeren Patienten ist zudem die Messung der Kalzium- und Phosphatkonzentration im 24h-Urin zum Ausschluss einer familiären hypokalziurische Hyperkalzämie (FHH) sinnvoll.
Insbesondere bei Vorhandensein von Osteolysen und im Raum stehendem Malignomverdacht ist an eine paraneoplastische ektope Sekretion PTH-verwandter Peptide (PTHrP) als Ursache der Hyperkalzämie zu denken und PTHrP zu bestimmen. Dies kann auch unabhängig von einer ossären Metastasierung auftreten und findet sich meist bei Bronchialkarzinomen. Aus diesem Grund sollte eine unklare Hyperkalzämie auch eine Tumorsuche nach sich ziehen. Im Fallbeispiel fand sich hierfür kein Anhalt (PTHrP < 0,50 pmol/l).
Tabelle 2 zeigt die typischen laborchemischen Konstellationen beim pHPT und seinen wichtigsten Differentialdiagnosen. Allerdings tritt diese nur in rund 80 - 90 Prozent der Fälle auf. Es kann bei begleitender Niereninsuffizienz, Albuminmangel oder simultanem Vitamin D-Mangel auch das Bild eines normokalzämen pHPT auftreten. Zudem muss auch bei normwertigem oder nur leicht erhöhtem PTH (10 - 20 % der Fälle) weiter an einen pHPT gedacht werden, da in diesen Fällen PTH relativ zur Hyperkalzämie dennoch zu hoch ist. Normal wäre in diesen Fällen eine PTH-Suppression.
| Tabelle 2: typische laborchemische Konstellationen beim pHPT und seine wichtigsten Differentialdiagnosen | |||||||
|---|---|---|---|---|---|---|---|
| Ca2+ (S) | Ca2+ (U) | PO43- (S) | PO43- (U) | PTH intakt | AP | PTHrp |
pHPT | ++ | ++/ nw | -- | ++ | ++ | ++ | -- |
sHPT | --/ nw | -- | --/ nw/ ++ |
| ++ | ++ | -- |
tHPT | nw |
|
|
| ++ | ++ | -- |
MAH | ++ | ++ | -- | ++ | -- | ++ | ++ |
FHH | ++ | -- |
|
| ++/ nw | nw | -- |
(nw = normwertig, A>P = alkalische Phosphatase, p / s / t HPT = primaärer / sekundärer / tertiärer HPäT, MAH = malignomassoziierte Hyperkalzämie)
Sobald die Diagnose eindeutig ist, muss eine Sonographie der Nebenschilddrüsen (10 MHz Schallkopf) mit Darstellung aller vier Epithelkörperchen erfolgen. Adenome kommen echoarm zur Darstellung. Sollte dies nicht erfolgreich sein, ist gegebenenfalls eine 99mTc-MiBi-Szintigraphie oder ein CT des Thorax anzuschließen, da die NSD manchmal auch ektop im Mediastinum liegen können (10 %).
Therapie
Die Operation ist die Therapiemethode der Wahl und sollte nur in spezialisierten Zentren durchgeführt werden. Als formal eindeutige Indikationen gelten hierbei jede Form des symptomatischen pHPT sowie eine asymptomatische Erkrankung bei Vorliegen eines oder mehrerer der folgenden Kriterien:
- Serumkalzium > 0,25 mmol/l über der Normgrenze
- GFR < 60 ml/min
- Abnahme der Knochendichte (T-Score < 2,5 oder pathologische Fraktur)
- Alter < 50 Jahre
- Begleitfaktoren die eine hyperkalzämische Krise begünstigen wie Bettlägerigkeit
Bei eindeutiger präoperativer Darstellung aller NSD kann die OP auch minimal invasiv durchgeführt werden, wie dies bei unserer Patientin mittels MIVAP (= minimal invasive Video-assistierte Entfernung der Nebenschilddrüsen) erfolgte. Ansonsten müssen intraoperativ alle Epithelkörperchen aufgesucht werden. Bei Hyperplasie aller NSD wird eine simultane autologe Transplantation einer halben NSD in den M. brachioradialis oder M. sternocleidomastoideus durchgeführt. Intraoperativ erfolgt ein regelmäßiges PTH-Monitoring. Wenn der Wert intraoperativ um > 50 Prozent abfällt, gilt die Operation als erfolgreich (primäre Erfolgsquote 95 - 98 %.). Im geschilderten Fallbeispiel fiel PTH während der OP von > 2.000 pg/ml auf rund 300 pg/ml und lag am Folgetag schon im Normbereich.
Nach der OP kommt es zur langsamen Rückbildung fast aller Symptome. Schwere ossäre Veränderungen, renale und kardiale Folgeschäden sind jedoch nicht umkehrbar.
Postoperativ kann es vor allem bei Patienten mit hoher Knochenbeteiligung zu passageren Kalzium-Mangelzuständen, unter Umständen mit Rekalzifizierungstetanien und den hierfür typischen Chvostek- und Trousseau-Zeichen kommen (sog. "hungry bone Syndrom“). Aus diesem Grund sind engmaschige Laborkontrollen und gegebenenfalls eine Kalziumsubstitution postoperativ obligat.
Bei fehlender Operabilität des Patienten sollte auf eine ausreichende Flüssigkeitszufuhr geachtet werden; eine kalziumarme Diät ist nicht nötig. Thiazide, Digitalispräparate und Lithium sind aufgrund ihrer kalziumretinierenden Wirkung absolut kontraindiziert! Insbesondere bei postmenopausalen Frauen kann die Gabe von Bisphosphonaten zur Osteoporoseprophylaxe erwogen werden. Alternativ kann durch das Kalziummimetikum Cinacalcet (Mimpara®) die Empfindlichkeit des Kalziumrezeptors an den Hauptzellen der NSD gesteigert und somit bremsend auf die PTH-Sekretion eingewirkt werden. Zudem wird der renale Kalziumrezeptor beeinflusst und so die Serumkalziumkonzentration vermindert. Allerdings gehen mit der Therapie häufig Übelkeit und Kopfschmerzen als auch hohe Kosten einher. In jedem Fall sind bei inoperablen Patienten regelmäßige Laborkontrollen und gegebenenfalls. auch Knochendichtemessungen indiziert.
Komplikationen
Eine hyperkalzäme Krise kann prinzipiell jederzeit ohne Vorboten spontan entstehen, droht aber insbesondere bei Kalziumwerten > 3,5 mmol/l und bei Hyperkalzämie-begünstigenden Faktoren (z.B. Immobilität, Thiazid-Therapie). Sie geht einher mit Erbrechen, Polyurie, Polydipsie und daraus resultierender Exsikkose sowie mit psychotischen Manifestationen bis hin zu Somnolenz und Koma. Es kann zum akuten Nierenversagen, zu Organkalzifizierungen und zum plötzlichen Tod durch Herzrhythmusstörungen kommen. Wichtigste therapeutische Maßnahme ist die forcierte Diurese mit 3 - 6 l 0,9 %-NaCl/die, gegebenenfalls begleitet von Furosemid-Infusionen (Wichtig: Kalium-Kontrolle!). Insbesondere bei tumorassoziierter Hyperkalzämie ist auch die Gabe von Bisphosphonaten indiziert; bei anderer Ursache ist dies „off-label“. Die Letalität der hyperkalzämen Krise liegt bei bis zu 50 Prozent.
Literatur
- Herold, G., et al., Innere Medizin, Köln (2012)
- Mutschler, E., et al., Arzneimittelwirkungen – Lehrbuch der Pharmakologie und Toxikologie, Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart (2008)
- Greten, H. et al., Innere Medizin, 13. Auflage, Thieme Verlag, Stuttgart (2010)
- Uptodate, www.uptodate.com, 34 Washington Street, Wellesley, Massachusetts 02481
Artikel zu:
- Primary hyperparathyreoidism: diagnosis, differential diagnosis and evaluation (05/2017)
- Primary hyperparathyreoidism: clinical manifestations (05/2017)
- Primary hyperparathyreoidism: management (05/2017)