Weiterführende Informationen und Differentialdiagnostik zur Zertifizierten Kasuistik "Kupferstoffwechselstörungen"
wissenschaftlich begutachtet durch Professor Dr. Malte Ludwig, Chefarzt der Abteilung Angiologie und Phlebologie der Interne Klinik Dr. Argirov, Berg.
von Beate Appenrodt und Tilman Sauerbruch
Inhalt
Pathogenese / -physiologie des M. Wilson
Abbildung 1
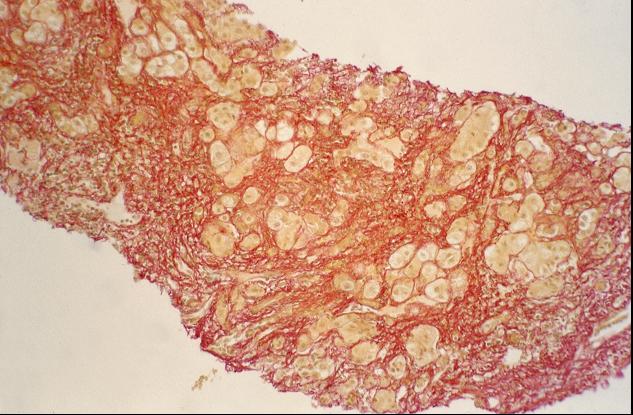
Die Abbildung 1: kleinknotige Leberzirrhose
Abbildung 2
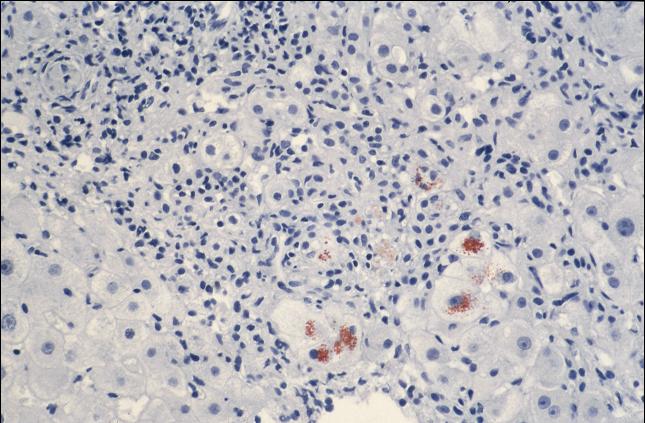
Abbildung 2: Pigmentablagerungen in der Rodaminfärbung.
Quelle: Professor Dr. Hans-Peter Fischer, Institut für Pathologie, Universitätsklinik Bonn
Definition des M. Wilson
Es handelt sich bei Morbus Wilson um eine seltene autosomal rezessiv vererbare Erkrankung des Kupfermetabolismus. Die Inzidenz beträgt 1:30000. Das Auftreten von heterozygoten Merkmalsträgern liegt bei 1:90 (1,2). Dabei manifestiert sich die hepatische Form des M. Wilson häufig vor dem 20. Lebensjahr, die neurologisch betonte Form der Erkrankung tritt eher um das 40. Lebensjahr auf mit einer großen Schwankungsbreite vom 5. bis zum 60. Lebensjahr.
Die Erkrankung ist durch eine Kupferanreicherung bzw. -überladung gekennzeichnet, die mit klinischen Folgen vor allem der Kupfer aufnehmenden Organe wie Leber, Basalganglien, Augen, der Nieren und des Blutes einhergeht. Daher ist auch die Bezeichnung "hepatolentikuläre Degeneration" für diese Erkrankung bekannt (Tabelle 1).
Erstmalig wurde ein neurologisches Syndrom mit Ataxie, Schwindel und Gangunsicherheit von S. Wilson 1912 beschrieben (3).
| Mögliche Befunde bei M. Wilson | |
|---|---|
| Hepatische Zeichen: | Transaminasenerhöhung Steatosis hepatis, Leberfibrose Hepato-/Splenomegalie Akute Hepatitis Leberzirrhose Akutes Leberversagen |
| Neurologische Zeichen: | Bewegungsstörung Tremor Dysarthrie Rigidität Ataxie Migräneattacken Schwindel Krampfanfälle Dyskinesie |
| Psychiatrische Zeichen: | Depression Neurosen (z.B. Angststörungen) Persönlichkeitsänderungen Affektive Störungen Psychosen |
| Blutbildveränderungen: | Microzytäre Anämie Hämolytische Anämie Leuko-/Thrombozytopenie |
| Augenveränderungen: | Kayser-Fleischer-Ring Sonnenblumenkatarakt |
Tabelle 1
Pathogenese/-physiologie des M. Wilson
Kupfer ist ein essentielles Spurenelement. Es laufen viele wichtige kupferhaltige Enzymreaktionen im Körper ab. Das tägliche Kupferangebot in der Nahrung liegt mit einer ungefähren Menge von ca. 1 - 3 mg deutlich über dem täglichen Bedarf.
Kupfer wird nahezu vollständig von den Hepatocyten aufgenommen. Dort wird es zum weiteren Transport im Körper an Coeruloplasmin gebunden. Das überschüssige, freie und stark zelltoxische Kupfer wird biliär ausgeschieden (2).
Der Gendefekt bei der Wilson-Krankheit liegt auf dem langen Arm des Chromosoms 13. Dieses Gen kodiert ein ATP-abhängiges Kupfertransportprotein namens ATP7B oder auch Wilson disease protein genannt, welches im Golgi-Apparat, insbesondere der Hepatocyten lokalisiert ist. Die in Europa am häufigsten auftretende Mutation ist die Mutation H1069Q im Exon 14 (4).
Die Funktion dieses Proteins besteht zum einen darin, den Transport von Kupfer in das Golgi-System zum Einbau in das noch Kupfer-freie-Apocoeruloplasmin zu gewährleisten und zum anderen darin, die biliäre Exkretion des überschüssigen, freien, toxischen Kupfers zu ermöglichen.
Das Wilson-Protein hat also eine Doppelfunktion: Es stellt Kupfer-beladenes Coeruloplasmin dem Organismus zur Verfügung und gleichzeitig regelt es die Exkretion von überschüssigem Kupfer.
Bei niedriger Kupfermenge in den Hepatocyten wird das mit Kupfer beladene Coeruloplasmin (Holocoeruloplasmin) über Vesikel aus der Leber ausgeschleust. Bei einer hohen Kupferkonzentration wird Kupfer zu dem Wilson-Protein ATP7B, das sich auch an den Mikrotubuli der Gallekanaculi befindet, zur biliären Exkretion transportiert.
Klinik des M. Wilson
Die Ursache für die verschiedenen Formen der klinischen Manifestation ist die Kupferüberladung in unterschiedlichen Geweben. Dabei treten häufig hepatische und neurologische Manifestationen, jedoch auch Veränderungen des Auges und des Blutbildes auf.
Hepatisch
Bei der hepatischen Manifestation wird zwischen der akuten und der chronischen Verlaufsform unterschieden.
Das akute Leberversagen betrifft mehrheitlich jüngere Patienten aus völliger Gesundheit. Der Verlauf ist oft letal, es sei denn, der Patient wird transplantiert.
Das akute Leberversagen ist oft mit einer hämolytischen, Coombs-negativen, normocytären Anämie vergesellschaftet. Es kommt durch freies, toxisches Kupfer, das vor allem beim Zerfall der Hepatocyten freigesetzt wird, zu einer Störung der Natrium-Kalium-ATPase der Erythrocyten mit Hämolyse. Eine hämolytische Anämie ist also ein Zeichen für einen schweren, akuten Verlauf des M. Wilson, der so genannten Wilson-Krise (5).
Bei der chronischen Verlaufsform werden auch Blutbildveränderungen beobachtet: Das Kupfer-beladenen und somit funktionsfähige Coeruloplasmin spielt eine entscheidende Rolle in der enzymatischen Reaktion der Ferroxidase und dem Eisenstoffwechsel. Bei funktionsunfähigem Coeruloplasmin als Cofaktor der Ferroxidase kann dieser bei M. Wilson gestört sein, so dass es zu einer mikrocytären Anämie im Sinne einer Eisenmangelanämie kommen kann.
Der chronisch hepatische Verlauf kann mit Transaminasenerhöhung, Ikterus und Allgemeinzustandsverschlechterung einhergehen. Wird keine Therapie begonnen, kann sich das Bild einer Leberzirrhose mit Zeichen der portalen Hypertension entwickeln.
Neurologisch
Die neurologischen Manifestationen sind vorwiegend extrapyramidaler Natur aufgrund von Kupferüberladung der Basalganglien. So sind Symptome wie Ataxie, Tremor, Dyskinesie, Dysarthrie aber auch Sprachschwierigkeiten und Mikrographie zu beobachten.
Die neurologische Symptomatik kann sich ohne adäquate Therapie zu einer deutlichen Intelligenzminderung und deutlichen Bewegungseinschränkungen durch Ataxie, Spastiken und schwerer Rigidität entwickeln. Epilepsien sind heutzutage bei adäquater Therapie deutlich seltener geworden.
In der bildgebenden Diagnostik sieht man insbesondere Veränderungen im Bereich des Ncl. dentatus, im Thalamus, in der Capsula interna und in der grauen Substanz.
Die psychiatrischen Symptome sind vor allem Persönlichkeitsveränderungen wie Stimmungsschwankungen (z.B. Wutanfälle, Reizbarkeit) und Konzentrationsschwäche. Depressive Episoden und Psychosen können ebenfalls auftreten.
Als Augenbeteiligung wird der nach den Entdeckern 1902/03 genannte Kayser-Fleischer Ring oft gesehen. Dabei handelt es sich um eine braun-rötliche ringförmige Kupferablagerung in der Descemet-Membran der Kornea. Dies kann im Rahmen der augenärztlichen Untersuchung mit der Spaltlampe diagnostiziert werden und gilt als pathognomisch für M. Wilson. Allerdings tritt der Kayser-Fleischer-Ring nicht bei jeder Wilson-Erkrankung auf. So zeigt sich ein deutlich geringeres Auftreten des Kornealringes bei führender hepatischer Manifestation im Gegensatz zu neurologisch führender Symptomatik; hier ist fast immer ein Ring zu diagnostizieren.
Der Kayser-Fleischer-Ring hat kaum Krankheitswert. Es kann allenfalls zu leichter Einschränkung des Sehvermögens kommen. Er kann allerdings zur Diagnosestellung hilfreich sein. Ein nachgewiesener Kayser-Fleischer-Ring ist nicht pathognomisch für M. Wilson, so kann er auch bei anderen cholestatischen Lebererkrankungen wie zum Beispiel der Primär biliären Zirrhose vorhanden sein.
Als weitere Augenbeteiligung wird die Ausbildung eines Katarakts durch Kupferablagerung in der Linse (daher auch die andere Bezeichnung der Erkrankung: Hepatolentikuläre Degeneration) beobachtet. Es zeigt sich eine sonnenblumenartige Form des Katarakts.
Diagnostik des M. Wilson
Bei den klinischen Zeichen der Leberzirrhose wie zum Beispiel Sklerenikterus, Spider naevi, Aszites oder auch des akuten Leberversagens muss immer auch an M. Wilson gedacht werden, vor allem wenn gleichzeitig extrapyramidale Symptome bestehen.
Weitere laborchemische Befunde können die Verdachtsdiagnose erhärten.
Dabei ist folgende Konstellation typisch:
- erhöhte Konzentration des freien Kupfers im Serum (> 50µg/dl)
- erniedrigte Konzentration des Coeruloplasmins im Serum (< 20 mg/dl)
- erhöhte Kupferausscheidung im Urin (> 100µg/24h)
Es kann jedoch auch zu normalen bis erhöhten Konzentrationen des Coeruloplasmins kommen; einerseits unter Östrogeneinfluss wie zum Beispiel während der Schwangerschaft, andererseits als Akute-Phase-Reaktion. Bei der Coeruloplasmin-Bestimmung im Serum kommt es bei 5 Prozent der Patienten zu falsch negativen Ergebnissen. Bei Säuglingen kann man diesen Parameter bei physiologisch erniedrigtem Coeruloplasmin nicht als diagnostischen Wert verwenden.
Die Kupferausscheidung im Urin größer 100 µg/24h erhärtet die Diagnose des M. Wilson.
Ein weiterer wichtiger diagnostischer Schritt ist die perkutane Leberpunktion. Eine stark erhöhte Kupferkonzentration im Leberparenchym von mehr als 250µg/g ist beweisend für M. Wilson. Mikroskopisch kann sich das Bild einer kleinknotigen Leberzirrhose und der Kupferablagerungen in den Hepatocyten nach Rhodamin-Färbung zeigen (Tabelle 2).
Da es sich bei M.Wilson um eine genetisch vererbbare Erkrankung handelt, stellt sich die Frage, ob genetische Untersuchungen für die Diagnosesicherung herangezogen werden können. Dabei sind jedoch genetische Tests bei über 200 funktionell wirksamen Mutationen des Wilson-Gens nur bei positiver Familienanamnese sinnvoll.
Diagnostik des M. Wilson
| ||||||||||
Tabelle 2
Therapie des M. Wilson
Eine Therapie muss nach Diagnosestellung, unabhängig von dem Schweregrad der Erkrankung immer begonnen und dauerhaft fortgesetzt werden (Tabelle 3).
Ihr Ziel ist die Entfernung des überschüssigen Kupfers aus den Kupfer-speichernden Organen.
Dabei ist auf eine kupferarme Diät zu achten. Der Schwerpunkt der Therapie ist medikamentös. Bei einer führenden hepatischen Symptomatik ist die Indikation zur Lebertransplantation - abhängig von dem Grad der Leberdekompensation - als einzig verbleibende Therapieoptionen zu evaluieren.
Zur medikamentösen Therapie stehen zum einen Chelatbildner wie Penicilliamin (Metalcaptase) oder Trientine (Triethylen-tetramin-Dihydrochlorid) zur Verfügung, die mit Kupfer einen Chelatkomplex bilden und seine Ausscheidung erhöhen oder Zink, welches die Kupferaufnahme deutlich verringert.
Penicillamin sollte in einer initialen Dosis von täglich 1g verteilt auf ein bis zwei Dosen verabreicht werden. Die Therapie gilt als erfolgreich bei einer normwertigen Kupferkonzentration im Serum und einer Kupferausscheidung im Urin von weniger als 500 µg/24h (6).
Die Gabe von Penicillamin kann jedoch mit einer ganzen Reihe von Nebenwirkungen einhergehen: Es kann unter anderem zu Hautausschlägen, Fieber, Lymphadenopathien aber auch zur Ausbildung eines nephrotischen Syndroms oder zu Autoimmunphänomene wie Lupus erythematodes kommen (7).
Diese Nebenwirkungen können auch Jahre nach Therapiebeginn noch auftreten; so dass gelegentlich ein Wechsel der Therapie notwendig ist. Man kann dann den Chelatbildner wechseln und mit Trientine, welches nebenwirkungsärmer ist, beginnen. Aber auch hier muss auf Nebenwirkungen wie Eisenmangelanämie und Kontaktdermatitis geachtet werden.
Die Dosis von Trientine liegt zwischen 750 bis 1250 mg täglich und sollte auf 2 - 4 Einnahmen oral täglich verteilt werden.
Eine weitere Therapieoption ist die Gabe von Zink. Zink wirkt zum einen über die Hemmung der Kupferabsorption durch kompetitive Hemmung der Kupfer-aufnehmenden Transporter an den Darmmukosa-Zellen und zum anderen über die Induktion des Kupfer-aufnehmenden Proteins Metallothionein, das Kupfer in den Mukosazellen des Darms speichern und unwirksam machen kann (8).
Zusammenfassend sollte die medikamentöse Therapie kombiniert mit einer kupferarmen Diät konsequent lebenslang durchgeführt werden. Dabei ist jedoch mit einem Therapieerfolg im Sinne eines Rückgangs der freien Kupferkonzentration im Serum und der Symptomatik erst circa ein halbes Jahr nach Therapiebeginn zu rechnen.
Wie oben erwähnt und im Falle der jungen Patientin, die in der Kasuistik beschrieben wurde, ist bei führender hepatischer Erkrankung mit Zeichen eines akuten Leberversagens oder einer dekompensierten Leberzirrhose die Indikation zur Lebertransplantation gegeben. Da vor allem die ATP7B-Expression in den Hepatocyten verantwortlich für die Krankheit ist, ist nach Lebertransplantation eine weitere medikamentöse Therapie in der Regel nicht mehr notwendig (9). In der Zukunft könnten auch gentherapeutische Ansätze in der Therapie des M.Wilson eine entscheidende Rolle spielen.
Man sollte bei unklarer hepatischer oder/und neurologischer Symptomatik bei jüngeren Patienten, jedoch auch bei Erwachsenen um die 40 Jahre bis sogar in Einzelfällen um die 60 Jahre, an die seltene Speichererkrankung denken. Mittels einfacher labrochemischer Untersuchungen und ggf. mit einer Leberhistologie kann die Diagnose gesichert werden. Eine sich daran anschließende konsequent durchgeführte, lebenslange Therapie sollte rasch erfolgen und kann den Krankheitsverlauf entscheidend beeinflussen, je früher sie begonnen wird.
| Medikamentöse Therapie des M. Wilson | ||
|---|---|---|
| Präparat/Wirkung | Dosierung | Nebenwirkungen |
| D-Penicillamin (Chelatbildner, Kuperausscheidung steigt) | 900-2400 mg/24h | Überempfindlicheitsreaktionen, Nephrotoxische Wirkung, Autoimmunphänomen |
| Trientine (Chelatbildner, Kuperausscheidung steigt) | 1200-2700 mg/24h | Eisenmangelanämie, Gastritis |
| Zink (-sulfat,-azetat,-histidin) (Hemmung der intestinalen Kupferabsorption) | 45 mg/24h (Zink) | Gastrointestinale Beschwerden, Dyspepsie, Erhöhung der Pankreasenzyme |
Tabelle 3
Literatur und Links
1) Schilsky M.L. Wilson disease: genetic basis of copper toxicity and natural history. Semin Liver Dis 1996; 16: 83 - 95.
2) Gitlin JD. Wilson disease Gastroenterology 2003; 125: 1868 - 1877.
3) Wilson S. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain 1912; 34: 295.
4) Riordian S., Williams R. The Wilson`s disease gene and phenotypic diversity. J Hepatol 2001; 34: 165 - 171.
5) Stremmel W. Diagnostic characteristicsof acute hepatic failure in fulminant Wilson`s disease Z Gastroenterol 1992; 61: 317 - 328.
6) Walshe JM. Penicillamine, a new oral therapy for Wilson`s disease. Am J Med 1956; 21: 487 - 495.
7) Walshe JM. Treatment of Wilson´s disease with trientine. Lancet 1982; 1: 643 - 647.
8) Oestreicher P., Cousins RJ. Copper and zinc absorption in the rat: mechanism of mutual antagonism J Nutr 1985; 115: 159 - 166.
9) Sternlieb I. Wilson`s disease: indications for liver transplants. Hepatology 1984; 4: 15 - 17.
Leitlinie zu M. Wilson der Deutschen Gesellschaft für Neurologie (2003)
American Association for the Study of Liver Diseases (AASLD)