Weiterführende Informationen und Differentialdiagnostik zur Zertifizierten Kasuistik: Blaue Zehen, blaue Finger
Folge 63 der Reihe Zertifizierte Kasuistik
von Nicole Bogun und Joachim Schrader
In der vorgestellten Kasuistik handelt es sich um ein paraneoplastisch aggraviertes Antiphospholipid-Syndrom (APLS) bei vorbekanntem primärem APLS.
Definition und Risikofaktoren bei Antiphospholipid-Syndrom (APLS)
Hierbei handelt es sich um eine systemische Autoimmunerkrankung mit arteriellen und / oder venösen Thrombosen und / oder geburtshilflichen Komplikationen bei Frauen mit gehäuften (Spät-)Aborten, Früh- oder Todgeburten bei Anwesenheit von Lupusantikoagulans und/oder Kardiolipin-Antikörpern IgM und IgG und / oder β2-Glykoprotein IgM und IgG (1). In der Gesamtbevölkerung sind Antikörper gegen Phospholipide zu 2 - 5 Prozent vorhanden, bei SLE-Patienten zu 30 - 40 Prozent (3) nachgewiesen, zu 25 Prozent bei Frauen mit rez. Aborten und Todgeburten. Es besteht aber auch ein gehäufter Nachweis von 10 - 20 Prozent im Patientengut bei unter 45-jährigen „young stroke“- Patienten oder frühem Myokardinfarkt (1,2,10).
Es wird ein primäres Antiphospholipid-Syndrom, das heißt ohne erkennbare Ursache oder Grunderkrankungen wie Autoimmun- oder Tumorerkrankungen unterschieden von einem sekundären APLS. Beim sekundären APLS dominiert zu 36 Prozent der systemische Lupus erythematodes sowie Mischkollagenosen. Es tritt auch bei rheumatoider Arthritis, M. Behcet, onkologischen Erkrankungen wie das MGUS und M. Waldenström und parainfektiös wie bei Hepatitis C, HIV, Q-fieber, Malaria auf (5,10).
Klinische Manifestationen
Neben venösen und arteriellen Thrombosen können Manifestationen des APLS das zentralnervöse Nervensystem betreffen und
- Kopfschmerzen,
- Migräne,
- juvenile Schlaganfälle und
- Demenz
auslösen.
Kardial wurden thrombotische Auflagerungen der Aorten- und Mitralklappe, Koronararterienverschlüsse und pulmonal-arterielle Hypertonie beschrieben. Als Hautmanifestationen finden sich die
- Livedo racemosa,
- Purpura,
- Ulcera und
- Gangräne.
Nephrologisch gibt es eine sogenannte APS-Nephropathie und Nierenvenenthrombosen. Weitere beschriebene Manifestationen sind Osteonekrosen, Mesenterialischämie, M. Addison und das Budd-Chiari-Syndrom (5, 6, 10).
Labor-Diagnostik
Antiphospholipid-Antikörper (aPL-AK) sind der Sammelbegriff für Lupus-Antikoagulans, Cardiolipin-Antikörper und β2-Glykoprotein. Diese drei sollten bei Verdacht auf ein APLS immer getestet werden. Das Lupus-Antikoagulans wird mittels Gerinnungstests bestimmt. Der Test ist spezifischer, wird aber durch alle Antikoagulanzien, insbesondere DOAKs gestört. Laborbestimmunen von Cardiolipin-AK und β2-Glykoprotein sind von der Gabe von Antikoagulanzien unabhängig. Sie werden mittels hochwertiger ELISA-Testsysteme nachgewiesen. APLS-Patienten können in einem, zwei oder drei Testverfahren positiv getestet werden. Daher sollten unbedingt alle Testverfahren durchgeführt werden. Besonders schwere Verläufe sind aber meistens triple-positiv. Nach 12 Wochen ist eine Bestätigung notwendig (1).
Die PTT-Bestimmung ist oft gestört, das heißt sie ist verlängert schon ohne Antikoagulation, obwohl eine prothrombogene Gerinnungssituation vorliegt.
Das APLS gilt als gesichert, wenn ein klinisches Kriterium und ein Laborkriterium erfüllt sind (1).
Therapie
Thrombosen beim APS werden leitliniengerecht mit NMH, Fondaparinux oder Vitamin-K-Antagonisten behandelt. Bei dieser Gerinnungsstörung empfiehlt sich eine verlängerte Therapiedauer. Das Rezidivrisiko liegt bei 80 - 90 Proeztn (10). Die Datenlage zur Therapie mit DOAKs umfasst bisher aber nur wenige Studien (3, 4). Zuletzt zeigten aktuelle Studien besonders unter triple-positiven Patienten (7, 11) ein erhöhtes Rezidivrisiko. Eine Therapie ist daher nicht indiziert. Unfraktioniertes Heparin ist wegen der häufig erhöhten PTT schlecht steuerbar.
Bei der Therapie mit Cumarinen muss beachtet werde, dass die Coagucheck Selbsttestung gestört sein kann (10).
Arterielle Komplikationen erfordern neben einem Vitamin-K-Antagonsiten gegebenenfalls auch einen Thrombozytenaggregationshemmer (10).
Bei Schwangerschaftskomplikationen wirkt sich die kombinierte Gabe von niedermolekularem Heparin und ASS günstig aus auf den Schwangerschaftserhalt (8, 9). Bei SLE bietet sich eine Hydroxychloroquintherapie und gegebenenfalls Rituximab oder Sirolimus als immunmodulierende Therapie an (10).
Bei katastrophalen APLS-Verläufen (0,8 % der Patienten) mit disseminierter Thrombenbildung in zahlreichen Gefäßen, mit Multiorganversagen und Thrombozytopenie (5, 10) werden Glukokorticoide, Plasmapherese und iv. Immunglobuline eingesetzt. Eine nachfolgend dauerhafte Antikoagulation und Plättchenhemmung sowie perioperative Plasmapheresen sind bei diesen Patienten zu beachten.
Erläuterung zum Verlauf des in der Kasuistik geschilderten Patienten
Die Prognose ist in der Mehrzahl der Fälle gut, meistens kann von einer Ausheilung ausgegangen werden (ca. 70 bis 80 %), mit eventueller Persistenz harmloser Rhythmusstörungen z.B. in Form von vermehrten Extrasystolen. In rund 15 bis 30 Prozent der Fälle werden chronische Verläufe mit der Entwicklung einer dilatativen Kardiomyopathie und konsekutiver Herzinsuffizienz beobachtet [1,2,3,6]. Relativ selten ist der Ausgang letal.
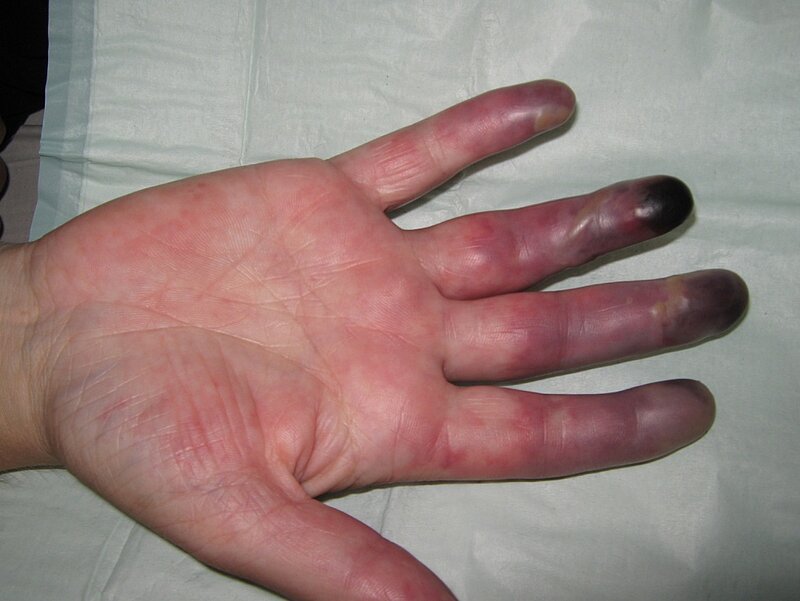
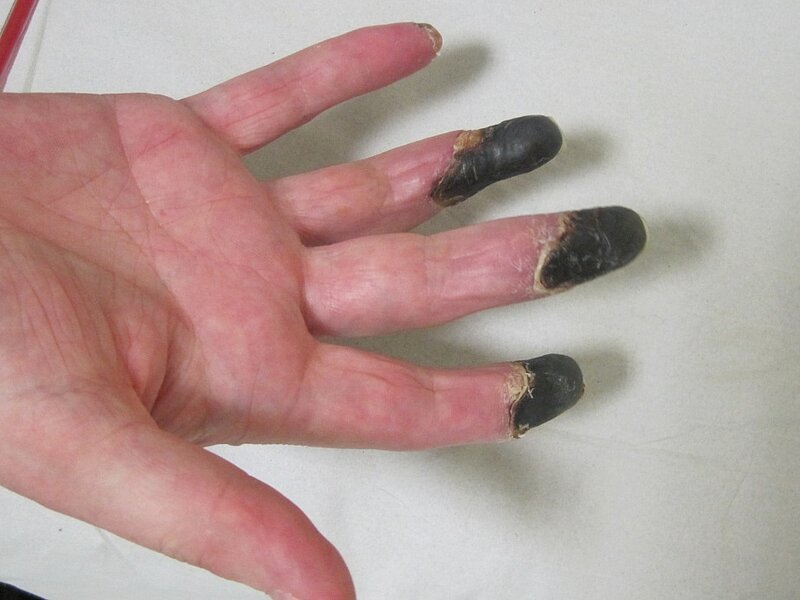
Der Patient erhielt eine sofort bei Aufnahme eingeleiteten iv. Prostanoidtherapie, eine niedermolekulare Heparintherapie in therapeutischer Dosierung sowie eine Wiedereinleitung der Acetylsalicylsäuregabe 100 mg und Cortikoidtherapie (1 mg/kg KG). Es kam darunter an den Akren von drei der vier Extremitäten nur zu einer leichten Verbesserung der schwer lividen Verfärbungen. An der rechten Hand entwickelten sich dagegen zunehmende schmerzhafte dunkellivide Verfärbungen (siehe Abbildung 1) bis hin zu Nekrosen der Endglieder Dig. 2 bis 5 innerhalb einiger Wochen (siehe Abbildung 2).
Eine Verlegung in eine universitäre hämatologische Spezialabteilung lehnte der Patient mehrfach ab. Als ultima ratio wurde bei erhöhten Fibrinogenwerten eine tägliche HELP-Apherese eingeleitet, um das erhöhte Fibrinogen zu senken. Erst darunter kam es zur Besserung des Lokalbefundes kranial der mittlerweile mumifizierten Finger Dig. 2 bis 5. Zusätzlich hatten sich oberflächliche Nekrosen Dig. 2 und 3 des rechten Fußes entwickelt.
Im Rahmen der umfangreichen Ursachenklärung fand sich kein Hinweis auf eine spezifische Kollagenose. Bei bereits vermuteter paraneoplastischer Genese fand sich in der Coloskopie ein hoch- bis mittelgradig differenziertes Adenocarcinom des Colon ascendens (pT3, pN0 (0/28), L0, V0, G2, R0, UICCStd. II). Dieses wurde operativ entfernt.
Postoperativ entwickelte der Patient starke vor allem nächtliche Ruheschmerzen der Zehen Dig. 2 und 3 rechts. Unterhalb der trockenen Zehenkuppennekrosen entwickelten sich neue livide Verfärbungen, eine Vorfußschwellung und ein vasculitisches Exanthem des Unterschenkel (siehe Abbildung 3). Eine erneute Umstellung des niedermolekularen Heparins auf ein orales Cumarin wurde daher eingeleitet. Zur Nachsorge blieb der Patient in engmaschiger ambulanter angiologischer Betreuung und erhielt lokale Wundtherapien und Antibiotika nach Abstrich und Resistenzprüfung.

Trotz der oben genannten Therapiemaßnahmen kam es zu einem progredienten Schub des Antiphospholipidsyndroms mit Anstieg der Cardiolipin-Ak-Titer. Daher willigte der Patient einer Verlegung in die Medzinische Hochschule Hannover (MHH) zur Plasmapherese und Immunglobulingabe ein. Darunter kam es zur Stabilisierung des Krankheitsbildes und die mittlerweile feuchten Nekrosen der Finger- und Zehenendglieder konnten durch die handchirurgische Abteilung der MHH amputiert werden.
Prä- und postoperativ erfolgten weitere Plasmapheresen und intravenöse Immunglobulingaben.
Zwei weitere Monate später konnte der Patient mit dem deutlich gebesserten Handbefund (siehe Abbildung 4) erfreulicherweise seinen Beruf wiederaufnehmen.

Literatur
- Miyakis S et al. J.Thromb Haemost. 2006;4(2), 295-306.
- Fischer M J et al. Arthr. Rheum.2007; 27(1),35-46
- Cohen H et al. Lupus 2015; 24, 1087-1094
- Cohen H et al. Lancet 2016; 3:e426-36
- Euro-Phospholipid-Project, Cervera et al. Lupus 2009;18(10):889-93
- Uthman et al., Ann Rheum Dis; 78(2):155-61
- Pengo V, et al. Blood; doi:10.1182/blood-2018-04-848333
- Bertsias GK et al. Ann Rheum Dis, 2008 ; 67(2):195-205
- Schreiber K, Hunt BJ Semin Throm Haemost 2016; 780-88
- Linnemann B, VASA 47(6):451-64
- Rote-Hand-Brief des BfArM, 23.5.19